

On-Demand Webinars
-

Using Linked-read Sequencing with Museums Collections for Studying Chromosome Evolution and Conservation Genomics in Birds
Taylor Hains
PhD Candidate | University of Chicago & The Field Museum
-
TELL-Seq users experience sharing Linked-read Sequencing with museum's collections for studying Chromosome Evolution and Conservation Genomics in birds.

-

Library Preparation and Genome Assembly Using Transposase Enzyme-Linked Long-Read Sequencing
Dr. Alvaro Hernandez
Director | DNA Services at the University of Illinois at Urbana Champaign
-
Dr. Hernandez discusses how his team sequenced and assembled the genomes of nine insects using Transposase Enzyme-Linked Long-Read Sequencing (TELL-Seq) at different stages of genome assembly. The unique contributions of TELL-Seq libraries to the completeness of the assembly, as well as the DNA extraction, library preparation, and sequencing methods the team employed, will be discussed in detail.

-
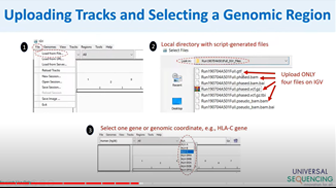
Visualization of Tell Sort data using the Integrative Genome Viewer
Dr. Yu Xia
Staff Bioinformatics Scientist | Universal Sequencing
-
Visualization of Tell-Sort data using the Integrative Genome Viewer (IGV) enhances the interpretation of phasing results generated with the TELL-Seq Library Prep kit developed by UST. Dr. Yu will describe how to find and run a script that converts Tell-Sort outputs into IGV-compatible files that conveniently display phasing data, and more.
